
内科学 第10版 「遺伝性脊髄小脳失調症」の解説
遺伝性脊髄小脳失調症(脊髄小脳変性症)
概念
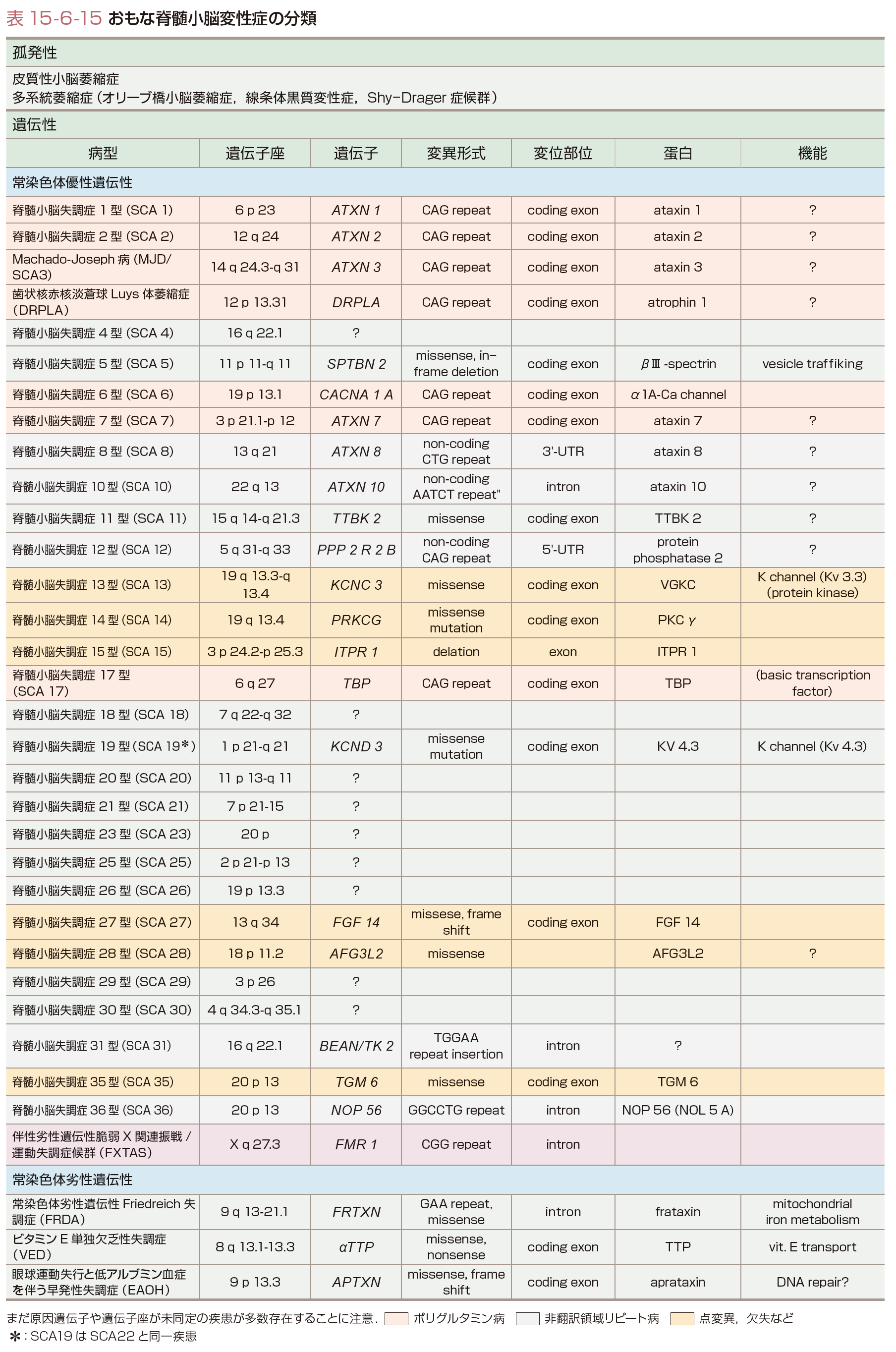
わが国の脊髄小脳変性症(spinocerebellar degeneration:SCD)のなかで遺伝性のものは約30%で,ほかの神経変性疾患と比べその割合が大きい.そのほとんどは常染色体優性遺伝性で,現在,約30と非常に多数の疾患が知られている(表15-6-15).SCDは人種差や地域差が大きく,わが国ではMachado-Joseph病(MJD)あるいは脊髄小脳失調症3型(spinocerebellar ataxia type 3:SCA 3),SCA 6,SCA 31,歯状核赤核淡蒼球Luys体萎縮症(dentatorubral-pallidoluysian atrophy:DRPLA)で大部分を占め,ほかの病型はまれである.常染色体劣性遺伝性では欧米人で最も多い遺伝性SCDであるFriedreich失調症が有名であるが,わが国には存在せず,低アルブミン血症を伴う早発性失調症あるいは眼球運動失行を伴う失調症1型,単独ビタミンE欠損症を伴う失調症などが少数みられるのみである.
病因・病態
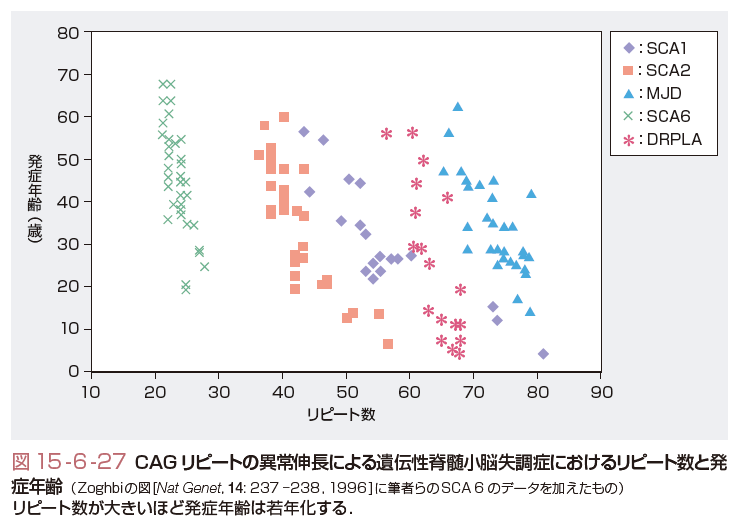
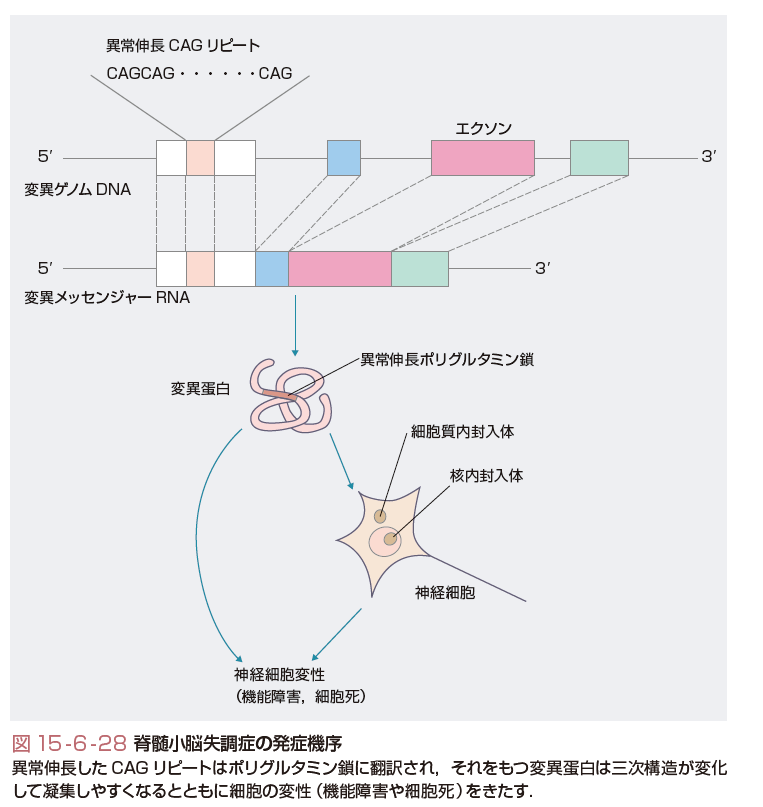
遺伝子変異としては,①エクソンのCAGの3塩基繰り返し配列(リピート)の異常伸長(ポリグルタミン病ともよばれる),②通常の点変異や欠失,③非翻訳領域のリピートの異常伸長に大別される(表15-6-15).①ではリピート数が大きいほど発症年齢が若く(図15-6-27),重症化する.世代が若返るごとに発症年齢が若くなる表現促進現象(anticipation)はCAGリピートが不安定で世代間で異常伸長することに対応している.変異蛋白は凝集しやすく,核内や細胞質内に凝集体あるいは封入体を形成するが,その過程で細胞機能を障害し最終的には神経細胞死をきたすと考えられる(図15-6-28,15-6-29).
神経症状・徴候
SCA 6,SCA 31などでは,ほぼ小脳失調症状(失調性歩行,四肢協調運動障害,書字障害,失調性構音障害,眼振)と筋トーヌス低下に終始し,MJD/SCA 3,SCA 1,SCA 2,DRPLAでは小脳系以外の系統も障害され,小脳症候に加えてパーキンソニズム,不随意運動,錐体路症候,自律神経症候,末梢神経症候などがさまざまな程度に,しかしながら一定の特徴をもった組み合わせで出現する.これらの症候は,一度に出現するわけではなく時間経過とともに加わってくることが多く,経過とともに単なる重症化ではなく病像そのものが変化することがある.
検査成績
最も重要なのは脳MRIであり,血管障害,炎症性疾患など二次性の小脳疾患の鑑別はもちろん,小脳・脳幹・大脳基底核・大脳皮質・大脳白質・脊髄などにつき萎縮の有無・程度と内部の信号強度の変化をチェックする.SCA 6では小脳虫部前方優位に小脳皮質の萎縮がみられ(図15-6-30),多系統障害型では脳幹の萎縮などが加わる.電気眼振図検査などで小脳性眼球運動障害を定量的に評価できる.その他,障害される神経系統に応じて神経伝導速度,誘発電位,筋電図などさまざまな検査で異常がみられることがあるが,血液,尿,髄液などの一般検査では普通異常はみられない.
診断・鑑別診断
家族歴が重要であるが,ときに家族歴がないこともあり,そのときは孤発性SCDとして,まず二次性小脳失調症を鑑別することが必要である.特徴的症候の組み合わせと経過により,各病型を臨床的にある程度診断することは可能であるが,正確を期するために遺伝子診断で遺伝子変異を証明することが望ましい.孤発性のときの遺伝子診断は陽性となったときの対応を十分に考えた上で慎重に判断する必要がある.
治療
まだ根本的治療は確立してはおらず対症療法と生活指導あるいはリハビリテーションが中心である.運動失調に対してはTRH製剤である酒石酸プロチレリン0.5~2.0 mgの筋注や静注,TRH誘導体のタルチレリン水和物の5 mg錠1日2回経口投与が有効なことがある.本症そのものが死因となることはまれであり,多くは臥床するようになってからの感染などにより死亡する.したがってできるかぎり寝たきりになるのを防ぐことが生命予後の観点からも重要であり,日常生活においては転倒防止などさまざまな工夫により自立した生活をできるかぎり持続させる努力が大切である.そのほか,パーキンソニズムには抗Parkinson病薬,痙性麻痺には抗痙縮薬,不随意運動にはそれぞれの対症療法薬を用いる.
おもな病型
1)SCA 1:
1974年わが国のYakuraらが初めて第6染色体に存在するHLAに連鎖することを報告し,後にataxin 1遺伝子のCAGリピートの異常伸長が原因と判明した.発症年齢は若年から中年と幅が広い.臨床病理学的には従来の遺伝性オリーブ橋小脳萎縮症に属し,小脳失調症候に加えて注視眼振,眼球運動制限,錐体路徴候,不随意運動などを呈するが,進行すると顔面・四肢筋の萎縮や腱反射低下も認められる.わが国では比較的まれである.
2)SCA 2:
ataxin 2遺伝子のCAGリピートの異常伸長により生じ,発症年齢は若年から中年と幅広い.臨床病理学的には従来の遺伝性オリーブ橋小脳萎縮症に属し,小脳失調症候に加えて眼球運動制限,緩徐眼球運動,振動覚低下,腱反射低下・消失,病的反射陽性,不随意運動(振戦,舞踏病様運動,ジストニア),人格変化,認知症,四肢筋萎縮などがみられる.特に眼球運動が非常に緩やかになる緩徐眼球運動が特徴的である.わが国では比較的まれである.
3)Machado-Joseph病(SCA 3):
優性遺伝性脊髄小脳失調症のなかで世界的に最も頻度が高い.わが国で遺伝子座・遺伝子が同定されたMJD1遺伝子のCAGリピートの異常伸長により生じる.若年から中年に幅広く発症する.小脳失調症が主体であるが顔面筋のミオキミア,びっくり眼,注視眼振,眼球運動制限,下肢痙縮,ジストニア・アテトーゼなどが特徴的である(図15-6-31).神経症候は多彩で錐体路・錐体外路徴候が中心のⅠ型,小脳徴候・錐体路徴候が中心のⅡ型,小脳徴候と筋萎縮・感覚障害・腱反射低下などがおもなⅢ型に分類される.神経病理学的にはおもに脊髄小脳路,橋小脳路,歯状核,赤核,淡蒼球,視床下核(Luys体),黒質,脊髄前角が障害されるが小脳皮質,大脳皮質,下オリーブ核は保たれるため,MRIでも小脳半球の萎縮は目立たず脳幹の萎縮が目立つ(図15-6-32).
4)SCA 6:
α1A電位依存性Caチャネル遺伝子(CACNA1A)のCAGリピートの4~18から(19)20~33への異常伸長によって生じる.臨床病理学的にはほぼ純粋な小脳失調症を呈する皮質性小脳萎縮症であり,わが国の遺伝性皮質性小脳萎縮症の約半数を占め,Machado-Joseph病についで多く,特に西日本に高頻度である.高齢発症で予後も比較的良好である.小脳性失調症状のほか,めまいの訴えや下眼瞼向き眼振が比較的多く,失調も含めときに反復発作性に現れることが特徴的である.同じ遺伝子の別の変異により反復発作性失調症2型(episodic ataxia type 2:EA2)や家族性片麻痺性片頭痛(familial hemiplegic migraine:FHM)が生じる.世代間や各組織間でリピート数がきわめて安定で,核内封入体はなくPurkinje細胞質内封入体がみられるなどほかのCAGリピート病と異なる特徴を有する.
5)SCA 7:
ataxin 7遺伝子のCAGリピートの異常伸長が原因である.臨床的には網膜黄斑部変性症を伴う遺伝性オリーブ橋小脳萎縮症であり,視力低下で発症することもあり注意が必要である.小脳失調症のほか,腱反射亢進,下肢の痙縮,緩徐眼球運動,外眼筋麻痺などを伴う.わが国では非常にまれである.
6)歯状核赤核淡蒼球Luys体萎縮症(DRPLA):
DRPLA遺伝子のCAGリピートの異常伸長が原因である.わが国に多く,わが国で記載され原因遺伝子も発見されたが,欧米では非常にまれである.表現促進現象が著明で発症年齢は小児から中年まで幅広く,発症年齢によって臨床症状が異なる.20歳未満の発症(若年型)ではミオクローヌス,てんかん,精神発達遅滞または認知症,小脳性運動失調症が主で,40歳以降の発症(遅発成人型)では小脳性運動失調症,舞踏病様不随意運動,性格変化,認知症が中心となり,20~40歳の発症(遅発成人型)では両者の中間の特徴を有する.眼振や錐体路徴候を呈することはあるが,外眼筋麻痺,筋萎縮,感覚障害などはみられない.脳MRIで小脳と脳幹の萎縮を認め,長期経過例では大脳白質にT2高信号域がみられることがある.尾状核の萎縮がみられない点がHuntington病との鑑別上重要である.
7)SCA 31:
第16染色体長腕にあるBEAN遺伝子のイントロンへのTGGAAの5塩基リピートの異常伸長の挿入変異により発症すると考えられている.SCA 6と同様に緩徐進行性のほぼ純粋な小脳失調症候,MRIでの小脳萎縮を呈するが,発症はより高齢である.[水澤英洋]
出典 内科学 第10版内科学 第10版について 情報